CENTER FOR THE SCIENCE OF
SYNTHESIS ACROSS SCALES
General-Purpose Peptoid High-Throughput Simulation Package
April 15, 2025
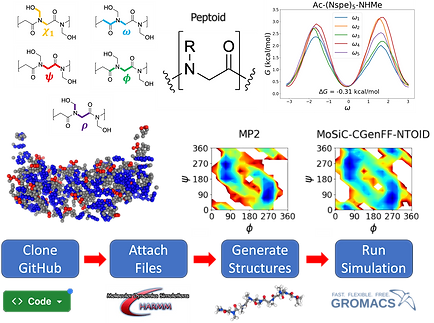
The MoSiC-CGenFF-NTOID simulation package enables high-throughput simulations of peptoids. It is accurate in describing states and transitions at the individual residue level, but can also explain self-assembly and aggregation phenomena.
Scientific Achievement
-
We developed the MoSiC-CGenFF-NTOID model tailored for accurate all-atom simulations of polypeptoids. This modular approach allows the incorporation of various side chain chemistries without the need for extensive reparameterization.
Significance and Impact
-
Implementation of host-guest interactions greatly streamlines the construction of artificial protein assemblies, opening new paths for designing proteins with novel structural, mechanical, and functional properties.
Research Details
-
Modular Framework: Separates backbone and side chain parameters, enabling reuse of existing CGenFF parameters.
-
Validation: Matches well with quantum calculations and experimental data, confirming its reliability.
-
Side Chain Library: Includes parameters for all 20 proteinogenic and 13 common synthetic side chains, extensible to new residues.
-
Resources: The force field and a structure-building tool are openly available to support community adoption.
Berlaga, A., K. Torkelson, A. Seal, J. Pfaendtner, and A.L. Ferguson. (2024). A Modular and Extensible CHARMM-Compatible Model for All-Atom Simulation of Polypeptoids. J Chem Phys, 161(24): 244901. https://doi.org/10.1063/5.0238570.
Work performed at the UChicago, University of Washington, and NC State University.